Ligand Design for Selective Metal Ion Recognition and Separation
The origins of this project in our group perhaps go back to my undergraduate studies at U.C. Berkeley where I first encountered coordination chemistry in the research laboratory of Prof. Dick Powell and where I made first contacts with actinide chemistry through the Radiation Laboratory on "the Hill." The interest sharpened during my postdoctoral studies at LANL (then LASL). Although no actinide chemistry was done by me in those early times at U.C. Berkeley, some of the research concepts were spawned there.
Our first efforts in the laboratory were stimulated at a small DOE meeting in Idaho in 1978. The DOE has the primary responsibility for the stewardship of the U.S. nuclear materials. As part of that responsibility, the DOE supports basic research in f-element chemistry at the National Laboratories and academic institutions. Some of that research activity is devoted to the discovery of new approaches for separation and detection of actinide ions in laboratory and large scale process settings. The cartoon presented here summarizes that important task. That is, there is need to develop "fishhooks" that can be used to retrieve selected f-element ions (fish) from nuclear materials process streams that contain complex mixtures of other ions (fish).

Given the conditions usually encountered with these process streams (highly acidic, highly basic, highly salted) this is no easy task. Furthermore as represented in the second cartoon below, separation alone is not sufficient. The f-elements species should be recovered for reuse in a variety of applications. In 1978, the novel extraction performance of carbamoyl methyl phosphonate (CMP) ligands were beginning to emerge. These ligands were found to do something no other ligand could do: selectively bind to lanthanide and actinide ions in strongly acidic acid solutions. The higher the acid strength, the stronger they bonded and the better the separation. Why? Why was the performance of this potentially bidentate chelating ligand so much better than its component phosphonate and amide parts? The questions posed at the Idaho meeting stimulated our 25+ year study of the coordination chemistry of lanthanide and actinide ions pertinent to practical separations and nuclear materials processing along with efforts to design functionalized organic ligands for actinide ion separations.

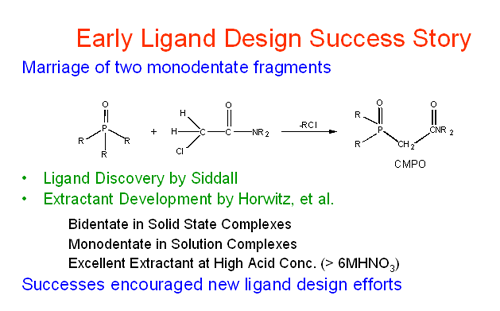
Our early studies first were directed toward characterization of the ligation behavior of the CMP ligands and their close relatives, carbamoyl methylphosphine oxides. As knowledge was gained we were led to examinations of modified phosphine oxides as shown here.
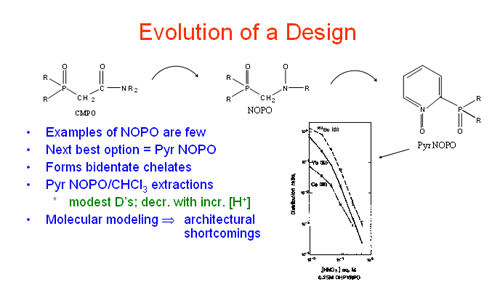
Most recently this project has focused on the design of new classes of ligands based upon the pyridine and pyridine N-oxide platforms. It was already known that pyridine N-oxide, on its own, is a hard donor, it readily forms coordination complexes with f-element ions and displays extraction performance related to TBP. Initially, we prepared ligands of type 1. It was thought that this compound would form energetically favored six-membered chelate rings. Indeed, stable coordination complexes with UO and Ln(III) ions were formed (Inorg. Chem.1987, 26, 1230; Inorg. Chem.1988, 27, 1220; Inorg. Chem.1988, 27, 3242). Subsequent liquid-liquid extractions (LLE) with a derivative of 1 showed that the ligand behaved more like TBP than CMP, showing larger distribution ratios (D) at low acid strengths (Radiochemica Acta 1989, 46, 123). Synthetic efforts then turned to the target ligand 2 where it was proposed that tridentate chelation might occur leading to stronger ligand binding. Despite considerable effort, only the bis-phosphine oxide could be isolated and no evidence for the N-oxide was found. In addition, a crystal structure determination for the pyridine compound (Inorg. Chim. Acta1986, 247, 29) and molecular modeling indicated that 2 might be sterically congested such that tridentate chelation on a Ln(III) ion might be energetically unfavorable. Relief of the steric crowding became the next goal.
 The next concept explored involved insertion of one or more methylene group spacers into the "donor arms" of 1 and 2. That is, 3 and 4 became the new target ligands. An initial synthetic scheme was developed and the coordination chemistry toward Ln(III), and Th(IV) was examined (Inorg. Chem.1993, 32, 2164). A most remarkable feature regarding these ligands became apparent from the structural studies. Despite adoption of seven-membered chelate rings, these ligands docked very comfortably on the highly polarizing Ln(III) and Th(IV) ions. Further, although electronically neutral, they displaced to the outer
The next concept explored involved insertion of one or more methylene group spacers into the "donor arms" of 1 and 2. That is, 3 and 4 became the new target ligands. An initial synthetic scheme was developed and the coordination chemistry toward Ln(III), and Th(IV) was examined (Inorg. Chem.1993, 32, 2164). A most remarkable feature regarding these ligands became apparent from the structural studies. Despite adoption of seven-membered chelate rings, these ligands docked very comfortably on the highly polarizing Ln(III) and Th(IV) ions. Further, although electronically neutral, they displaced to the outer 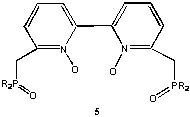 coordination sphere all or part of the counter anions (Cl - or ) present with the metal cations. This behavior by a neutral ligand toward Ln(III)/Th(IV) ions was quite unique. Subsequent studies with a bipyridyl N-oxide derivative 5 showed complete displacement of anions from Ln(III) ions and tetradentate ligand coordination.
coordination sphere all or part of the counter anions (Cl - or ) present with the metal cations. This behavior by a neutral ligand toward Ln(III)/Th(IV) ions was quite unique. Subsequent studies with a bipyridyl N-oxide derivative 5 showed complete displacement of anions from Ln(III) ions and tetradentate ligand coordination.
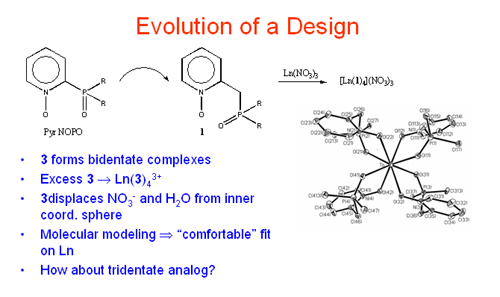
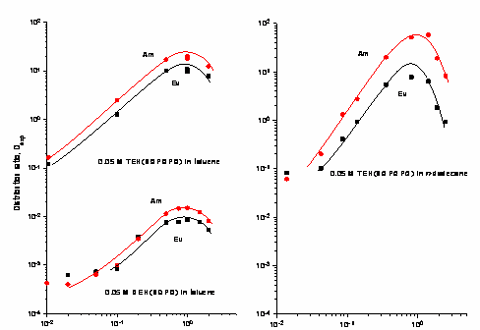
It was anticipated, based upon the results of the coordination chemistry, that extractions with 3 – 5 would be revealing. Indeed, initial studies using CHCl3 solutions of 4 toward acidic aqueous solutions containing Eu(III) and Am(III) proved to be very exciting (Solv. Extr. Ion Exch. 1997, 15, 381; Solv. Extr. Ion Exch. 1998, 16, 967; J. Alloys and Compounds. 1998, 271-273, 172). The ligand displayed favorable extraction properties similar to those found with the improved, carbamoylmethyl phosphine oxides (CMPO) where D values increased with increasing acid strength up to ~ 5M HNO3.
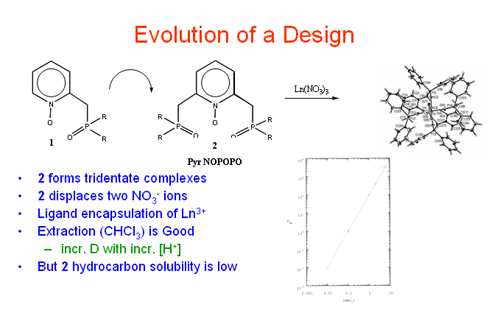
These favorable results have led to more expansive studies of the synthesis of 3 and 4, additional coordination chemistry and more through extraction analyses. Highlights include the following. In order to make ligands such as 3 and 4 practical in LLE separations, derivatives having good solubility and phase performance in hydrocarbon solvents are required. Therefore, a detailed synthetic effort to prepare derivatives with "greasy" substituents was undertaken (Inorg. Chem. 2001, 40, 4420). The successful completion of that work allowed for a thorough study of the thermodynamics of LLE of 3 and 4 toward Eu(III) and Am(III) (Inorg. Chem. 2002, 41, 5849) and that work confirms that 4 in particular, offers exciting potential for a practical, large-scale, extractant in advanced nuclear waste separations schemes. That activity occupies part of our attention at the current time.
The group has also been active in related areas. In particular, we have collaborated with Dr. M. Neu at LANL to study the plutonium coordination chemistry of 4, 6 and 7. It has been found that 4 forms a complex with 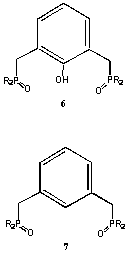 Pu(NO 3) 4 that is isostructural with the Th(NO 3) 4 complex. In each case two ligands bind in a tridentate chelate mode and displace two of the four nitrate counter ions to the outer coordination sphere (Inorg. Chem.2000, 39, 4152). The one-armed ligand 3 unexpectedly does not form tetrakis complexes as found with Ln(III) ions. Instead, an interesting bis-complex, [Pu(NO 3) 3[Pu(NO 3)] 0.5 is formed (J. Chem. Soc. Dalton2002, 2328).
Pu(NO 3) 4 that is isostructural with the Th(NO 3) 4 complex. In each case two ligands bind in a tridentate chelate mode and displace two of the four nitrate counter ions to the outer coordination sphere (Inorg. Chem.2000, 39, 4152). The one-armed ligand 3 unexpectedly does not form tetrakis complexes as found with Ln(III) ions. Instead, an interesting bis-complex, [Pu(NO 3) 3[Pu(NO 3)] 0.5 is formed (J. Chem. Soc. Dalton2002, 2328).
The group has also been exploring avenues to "soften" the donor field provided by 3 and 4. That effort includes the formation of thiophosporyl derivatives and the initial results of that work has appeared (J. Chem. Soc. Dalton2003, 4704). Most recently, the group has been developing syntheses for amide modified derivatives of 3 and 4 and these results will begin to appear in 2006.





